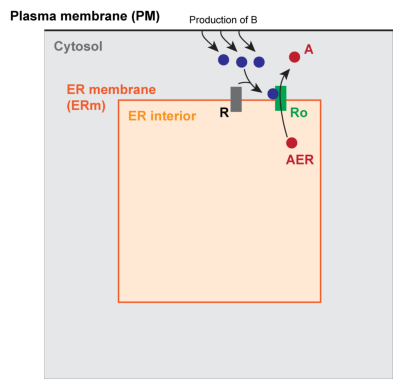
Example 5: Generic cell signaling system in 3D#
Geometry is divided into 4 domains; two volumes, and two surfaces:
cytosol (Cyto): \(\Omega_{Cyto}\)
endoplasmic reticulum volume (ER): \(\Omega_{ER}\)
plasma membrane (PM): \(\Gamma_{PM}\)
ER membrane (ERm): \(\Gamma_{ERm}\)
For simplicity, here we consider a “cube-within-a-cube” geometry, in which the smaller inner cube represents a section of ER and one face of the outer cube (\(x=0\)) represents the PM. The other faces of the outer cube are treated as no flux boundaries. The space outside the inner cube but inside the outer cube is classified as cytosol.
There are three function-spaces on these three domains:
In words, this says that species A and B reside in the cytosolic volume, species AER corresponds to an amount of species A that lives in the ER volume, and species R (closed receptor/channel) and Ro (open receptor/channel) reside on the ER membrane.
In this model, species B reacts with a receptor/channel, R, on the ER membrane, causing it to open (change state from R->Ro), allowing species A to flow out of the ER and into the cytosol. Note that this is roughly similar to an IP3 pulse at the PM, leading to Ca2+ release from the ER, where, by analogy, species B is similar to IP3 and species A is similar to Ca2+. A more comprehensive model of Ca2+ dynamics in particular is implemented in Example 6.
from matplotlib import pyplot as plt
import matplotlib.image as mpimg
img_A = mpimg.imread('example5-diagram.png')
plt.imshow(img_A)
plt.axis('off')
(-0.5, 2880.5, 2782.5, -0.5)
As specified in our mathematical documentation, assuming diffusive transport, the PDE and boundary condition for each of these volumetric species takes the form:
and the surface species take the form:
Our reaction terms and boundary conditions are chosen according to the system described above. For the purposes of this simplified example we use linear mass action in all reaction terms. Explicitly writing out this system of PDEs, we have:
Code imports and initialization#
import logging
import dolfin as d
import sympy as sym
import numpy as np
import pathlib
import gmsh # must be imported before pyvista if dolfin is imported first
from smart import config, mesh, model, mesh_tools, visualization
from smart.model_assembly import (
Compartment,
Parameter,
Reaction,
Species,
SpeciesContainer,
ParameterContainer,
CompartmentContainer,
ReactionContainer,
)
from smart.units import unit
/usr/lib/python3/dist-packages/scipy/__init__.py:146: UserWarning: A NumPy version >=1.17.3 and <1.25.0 is required for this version of SciPy (detected version 1.26.4
warnings.warn(f"A NumPy version >={np_minversion} and <{np_maxversion}"
We will set the logging level to INFO. This will display some output during the simulation. If you want to get even more output you could set the logging level to DEBUG.
logger = logging.getLogger("smart")
logger.setLevel(logging.INFO)
Futhermore, you could also save the logs to a file by attaching a file handler to the logger as follows.
file_handler = logging.FileHandler("filename.log")
file_handler.setFormatter(logging.Formatter(smart.config.base_format))
logger.addHandler(file_handler)
First, we define the various units for the inputs
# Aliases - base units
uM = unit.uM
um = unit.um
molecule = unit.molecule
sec = unit.sec
# Aliases - units used in model
D_unit = um**2 / sec
flux_unit = molecule / (um**2 * sec)
vol_unit = uM
surf_unit = molecule / um**2
Generate model#
Next we generate the model described in the equations above.
Compartments#
As described above, the three compartments are the cytosol (“Cyto”), the plasma membrane (“PM”), the ER membrane (“ERm”), and the ER interior volume (“ER”).
Note that, as shown, we can also specify nonadjacency for compartments; this is not strictly necessary, but will generally speed up the simulations.
Cyto = Compartment("Cyto", 3, um, 1)
PM = Compartment("PM", 2, um, 10)
ER = Compartment("ER", 3, um, 2)
ERm = Compartment("ERm", 2, um, 12)
PM.specify_nonadjacency(['ERm', 'ER'])
ERm.specify_nonadjacency(['PM'])
Initialize a compartment container and add the 4 compartments to it.
cc = CompartmentContainer()
cc.add([ERm, ER, PM, Cyto])
Species#
In this case, we have 5 species across 3 different compartments. We define each in turn:
A = Species("A", 0.01, vol_unit, 1.0, D_unit, "Cyto")
B = Species("B", 0.0, vol_unit, 1.0, D_unit, "Cyto")
AER = Species("AER", 200.0, vol_unit, 5.0, D_unit, "ER")
# Create an algebraic expression to define the initial condition of R
Rinit = "(sin(40*y) + cos(40*z) + sin(40*x) + 3) * (y-x)**2"
R1 = Species("R1", Rinit, surf_unit, 0.02, D_unit, "ERm")
R1o = Species("R1o", 0.0, surf_unit, 0.02, D_unit, "ERm")
Create species container and add the 5 species objects to it.
sc = SpeciesContainer()
sc.add([R1o, R1, AER, B, A])
Parameters and Reactions#
Parameters and reactions are generally defined together, although the order does not strictly matter. We define them in turn as follows:
# Degradation of B in the cytosol
k2f = Parameter("k2f", 10, 1 / sec)
r2 = Reaction(
"r2", ["B"], [], param_map={"on": "k2f"}, reaction_type="mass_action_forward"
)
# Activating receptors on ERm with B
k3f = Parameter("k3f", 100, 1 / (uM * sec))
k3r = Parameter("k3r", 100, 1 / sec)
r3 = Reaction("r3", ["B", "R1"], ["R1o"], {"on": "k3f", "off": "k3r"})
# Release of A from ERm to cytosol
k4Vmax = Parameter("k4Vmax", 2000, 1 / (uM * sec))
r4 = Reaction(
"r4",
["AER"],
["A"],
param_map={"Vmax": "k4Vmax"},
species_map={"R1o": "R1o", "uER": "AER", "u": "A"},
eqn_f_str="Vmax*R1o*(uER-u)",
)
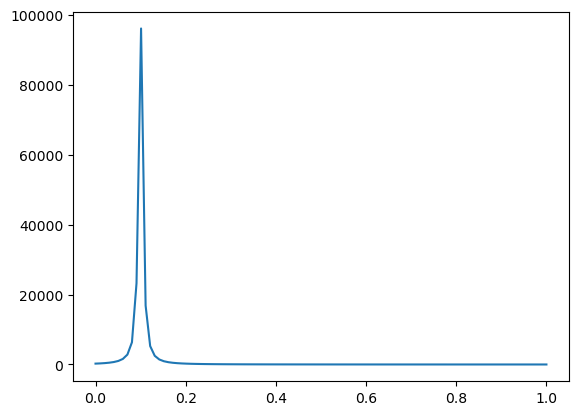
We define one additional reaction as the time-dependent production of species B at the plasma membrane. In this case, we define a pulse-type function as the derivative of an arctan function. Note that this is useful because we can provide an expression to use for pre-integration.
Vmax, t0, m = 500, 0.1, 200
t = sym.symbols("t")
pulseI = Vmax * sym.atan(m * (t - t0))
pulse = sym.diff(pulseI, t)
j1pulse = Parameter.from_expression(
"j1pulse", pulse, flux_unit, use_preintegration=True, preint_sym_expr=pulseI
)
r1 = Reaction(
"r1",
[],
["B"],
param_map={"J": "j1pulse"},
eqn_f_str="J",
explicit_restriction_to_domain="PM",
)
We can plot the time-dependent input by converting the sympy expression to a numpy function using lambdify.
from sympy.utilities.lambdify import lambdify
pulse_func = lambdify(t, pulse, 'numpy') # returns a numpy-ready function
tArray = np.linspace(0, 1, 100)
pulse_vals = pulse_func(tArray)
plt.plot(tArray, pulse_vals)
[<matplotlib.lines.Line2D at 0x7f23dd4bce80>]
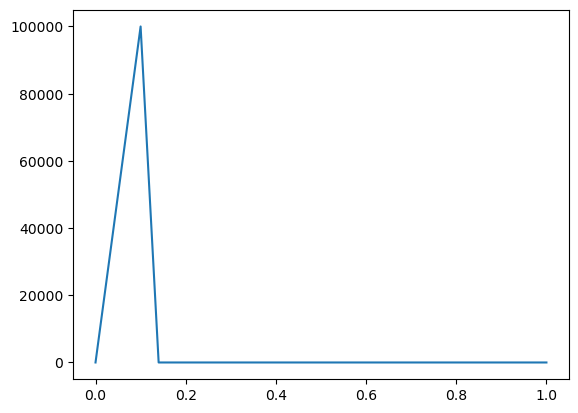
Note that we can also create a time-dependent parameter by reading data from a csv file. This is illustrated below, introducing a new parameter, “j1pulse_fromfile”, which we do not use in the full model in this case. It could be used in the model by simply defining it as “j1pulse” to overwrite the parameter previously defined from an expression.
j1pulse_fromfile = Parameter.from_file("j1pulse", "sample_input.csv", flux_unit)
tArray = j1pulse_fromfile.sampling_data[:, 0]
pulse_vals = j1pulse_fromfile.sampling_data[:, 1]
plt.plot(tArray, pulse_vals)
2026-03-05 08:21:52,479 smart.model_assembly - INFO - Loading in data for parameter j1pulse (model_assembly.py:646)
2026-03-05 08:21:52,480 smart.model_assembly - INFO - Time-dependent parameter j1pulse loaded from file. (model_assembly.py:678)
[<matplotlib.lines.Line2D at 0x7f23dd3403d0>]
We could also create a space- and time-dependent parameter by reading data from an xdmf file. This is illustrated below, introducing a new parameter, “j1pulse_fromxdmf”, which we use in an additional reaction that can be turned on or off below.
j1pulse_fromxdmf = Parameter.from_xdmf("j1pulse_fromxdmf", "test_data.xdmf", flux_unit, compartment = "PM")
add_reaction = True
if add_reaction:
jmag = Parameter("jmag", 0.0, unit.dimensionless)
r1add = Reaction(
"r1add",
[],
["B"],
param_map={"J": "j1pulse_fromxdmf", "mag": "jmag"},
eqn_f_str="mag*J",
explicit_restriction_to_domain="PM",
)
2026-03-05 08:21:52,557 smart.model_assembly - INFO - Loading in data for parameter j1pulse_fromxdmf (model_assembly.py:703)
2026-03-05 08:21:52,558 smart.model_assembly - INFO - Parameter j1pulse_fromxdmf linked to xdmf file. (model_assembly.py:727)
Create containers for parameters and reactions and add all the parameters and reaction objects to them.
pc = ParameterContainer()
rc = ReactionContainer()
if add_reaction:
pc.add([k4Vmax, k3r, k3f, k2f, j1pulse, j1pulse_fromxdmf, jmag])
rc.add([r1, r2, r3, r4, r1add])
else:
pc.add([k4Vmax, k3r, k3f, k2f, j1pulse])
rc.add([r1, r2, r3, r4])
Create/load in mesh#
In SMART we have different levels of meshes:
Parent mesh: contains the entire geometry of the problem, including all surfaces and volumes
Child meshes: submeshes (sections of the parent mesh) associated with individual compartments. Here, the child meshes are:
Cyto: the portion of the outer cube outside of the inner cube, defined by
cell_markers = 1ER: the inside portion of the inner cube, defined by
cell_markers = 2PM: surface mesh where x=0, defined by
facet_markers = 10ERm: surface mesh corresponding to all faces of the inner cube, defined by
facet_markers = 12
Here we create a UnitCube mesh as the Parent mesh, defined by
and we display a cut cross-section using pyvista.
domain, facet_markers, cell_markers = mesh_tools.create_cubes()
visualization.plot_dolfin_mesh(domain, cell_markers, clip_plane=(1,
1, 0), clip_origin=(0.5, 0.5, 0.5))
/usr/lib/python3/dist-packages/smart/visualization.py:283: PyVistaDeprecationWarning: This function is deprecated and will be removed in future version of PyVista. Use vtk with osmesa instead.
pyvista.start_xvfb()
ROOT -2026-03-05 08:21:57,709 trame_server.utils.namespace - INFO - Translator(prefix=None) (namespace.py:65)
ROOT -2026-03-05 08:21:57,752 wslink.backends.aiohttp - INFO - awaiting runner setup (__init__.py:147)
ROOT -2026-03-05 08:21:57,752 wslink.backends.aiohttp - INFO - awaiting site startup (__init__.py:154)
ROOT -2026-03-05 08:21:57,754 wslink.backends.aiohttp - INFO - Print WSLINK_READY_MSG (__init__.py:160)
ROOT -2026-03-05 08:21:57,754 wslink.backends.aiohttp - INFO - Schedule auto shutdown with timeout 0 (__init__.py:166)
ROOT -2026-03-05 08:21:57,755 wslink.backends.aiohttp - INFO - awaiting running future (__init__.py:169)
By default, smart.mesh_tools.create_cubes marks all faces of the outer cube as “10”, our marker value associated with PM. Here, since we are only treating the x=0 face as PM, we alter the facet markers on all other faces, setting them equal to zero. They are then treated as no-flux boundaries not belonging to a designated surface compartment. The resultant mesh with the new facet and volume markers is displayed below.
for face in d.faces(domain):
if face.midpoint().x() > d.DOLFIN_EPS and facet_markers[face] == 10:
facet_markers[face] = 0
img_mesh = mpimg.imread('example5-mesh.png')
plt.imshow(img_mesh)
plt.axis('off')
(-0.5, 1126.5, 738.5, -0.5)

We now save the mesh as an h5 file and then read it into SMART as a ParentMesh object.
mesh_folder = pathlib.Path("mesh")
mesh_folder.mkdir(exist_ok=True)
mesh_path = mesh_folder / "DemoCuboidsMesh.h5"
mesh_tools.write_mesh(
domain, facet_markers, cell_markers, filename=mesh_path
)
parent_mesh = mesh.ParentMesh(
mesh_filename=str(mesh_path),
mesh_filetype="hdf5",
name="parent_mesh",
)
2026-03-05 08:21:58,484 smart.mesh - INFO - HDF5 mesh, "parent_mesh", successfully loaded from file: mesh/DemoCuboidsMesh.h5! (mesh.py:245)
2026-03-05 08:21:58,485 smart.mesh - INFO - 0 subdomains successfully loaded from file: mesh/DemoCuboidsMesh.h5! (mesh.py:258)
Model and solver initialization#
Now we are ready to set up the model. First we load the default configurations and set some configurations for the current solver.
conf = config.Config()
conf.solver.update(
{
"final_t": 1,
"initial_dt": 0.01,
"time_precision": 6,
}
)
We create a model using the different containers and the parent mesh. For later reference, we save the model information as a pickle file.
model_cur = model.Model(pc, sc, cc, rc, conf, parent_mesh)
model_cur.to_pickle('model_cur.pkl')
Note that we could later load the model information from the pickle file using the line:
model_cur = model.from_pickle(model_cur.pkl)
Next we need to initialize the model and solver.
model_cur.initialize()
2026-03-05 08:22:03,511 smart.model - INFO - J01 = dF[ERm])/du[ER] is empty (model.py:1426)
2026-03-05 08:22:07,318 smart.solvers - INFO - Jpetsc_nest assembled, size = (10355, 10355) (solvers.py:201)
2026-03-05 08:22:07,319 smart.solvers - INFO - Initializing block residual vector (solvers.py:209)
2026-03-05 08:22:07,416 smart.model_assembly - INFO -
╒══════════════════╤════════════════════════════════════════════════╤═══════════════╕
│ │ Value/Equation │ Description │
╞══════════════════╪════════════════════════════════════════════════╪═══════════════╡
│ k4Vmax │ 2.00×10³ 1/µM/s │ │
├──────────────────┼────────────────────────────────────────────────┼───────────────┤
│ k3r │ 1.00×10² 1/s │ │
├──────────────────┼────────────────────────────────────────────────┼───────────────┤
│ k3f │ 1.00×10² 1/µM/s │ │
├──────────────────┼────────────────────────────────────────────────┼───────────────┤
│ k2f │ 1.00×10¹ 1/s │ │
├──────────────────┼────────────────────────────────────────────────┼───────────────┤
│ j1pulse │ 100000/(40000*(t - 0.1)**2 + 1) molecule/µm²/s │ │
├──────────────────┼────────────────────────────────────────────────┼───────────────┤
│ j1pulse_fromxdmf │ nan │ │
├──────────────────┼────────────────────────────────────────────────┼───────────────┤
│ jmag │ 0.00×10⁰ │ │
╘══════════════════╧════════════════════════════════════════════════╧═══════════════╛
(model_assembly.py:371)
2026-03-05 08:22:07,424 smart.model_assembly - INFO -
╒═════╤═══════════════╤═════════════════╤═════════════════════════════════════════════════════════════════╕
│ │ Compartment │ D │ Initial condition │
╞═════╪═══════════════╪═════════════════╪═════════════════════════════════════════════════════════════════╡
│ R1o │ ERm │ 2.00×10⁻² µm²/s │ 0.00×10⁰ molecule/µm² │
├─────┼───────────────┼─────────────────┼─────────────────────────────────────────────────────────────────┤
│ R1 │ ERm │ 2.00×10⁻² µm²/s │ (sin(40*y) + cos(40*z) + sin(40*x) + 3) * (y-x)**2 molecule/µm² │
├─────┼───────────────┼─────────────────┼─────────────────────────────────────────────────────────────────┤
│ AER │ ER │ 5.00×10⁰ µm²/s │ 2.00×10² µM │
├─────┼───────────────┼─────────────────┼─────────────────────────────────────────────────────────────────┤
│ B │ Cyto │ 1.00×10⁰ µm²/s │ 0.00×10⁰ µM │
├─────┼───────────────┼─────────────────┼─────────────────────────────────────────────────────────────────┤
│ A │ Cyto │ 1.00×10⁰ µm²/s │ 1.00×10⁻² µM │
╘═════╧═══════════════╧═════════════════╧═════════════════════════════════════════════════════════════════╛
(model_assembly.py:371)
2026-03-05 08:22:07,434 smart.model_assembly - INFO -
╒══════╤══════════════════╤═══════════╤════════════╤═════════╤════════════════╤═══════════════╕
│ │ Dimensionality │ Species │ Vertices │ Cells │ Marker value │ Size │
╞══════╪══════════════════╪═══════════╪════════════╪═════════╪════════════════╪═══════════════╡
│ ERm │ 2 │ 2 │ 218 │ 432 │ 12 │ 8.44×10⁻¹ µm² │
├──────┼──────────────────┼───────────┼────────────┼─────────┼────────────────┼───────────────┤
│ ER │ 3 │ 1 │ 343 │ 1296 │ 2 │ 5.27×10⁻² µm³ │
├──────┼──────────────────┼───────────┼────────────┼─────────┼────────────────┼───────────────┤
│ PM │ 2 │ 0 │ 289 │ 512 │ 10 │ 1.00×10⁰ µm² │
├──────┼──────────────────┼───────────┼────────────┼─────────┼────────────────┼───────────────┤
│ Cyto │ 3 │ 2 │ 4788 │ 23280 │ 1 │ 9.47×10⁻¹ µm³ │
╘══════╧══════════════════╧═══════════╧════════════╧═════════╧════════════════╧═══════════════╛
(model_assembly.py:371)
2026-03-05 08:22:07,442 smart.model_assembly - INFO -
╒═══════╤═════════════╤════════════╤═══════════════════════╤═══════════════════════╕
│ │ Reactants │ Products │ Equation │ Type │
╞═══════╪═════════════╪════════════╪═══════════════════════╪═══════════════════════╡
│ r1 │ [] │ ['B'] │ j1pulse │ volume_surface │
├───────┼─────────────┼────────────┼───────────────────────┼───────────────────────┤
│ r2 │ ['B'] │ [] │ k2f*B │ volume │
├───────┼─────────────┼────────────┼───────────────────────┼───────────────────────┤
│ r3 │ ['B', 'R1'] │ ['R1o'] │ k3f*B*R1-k3r*R1o │ volume_surface │
├───────┼─────────────┼────────────┼───────────────────────┼───────────────────────┤
│ r4 │ ['AER'] │ ['A'] │ R1o*k4Vmax*(-A + AER) │ volume_surface_volume │
├───────┼─────────────┼────────────┼───────────────────────┼───────────────────────┤
│ r1add │ [] │ ['B'] │ j1pulse_fromxdmf*jmag │ volume_surface │
╘═══════╧═════════════╧════════════╧═══════════════════════╧═══════════════════════╛
(model_assembly.py:371)
╒════╤═════════════╤══════╤══════════════════╤═════════════╤══════════════╤════════════════╕
│ │ name │ id │ dimensionality │ num_cells │ num_facets │ num_vertices │
╞════╪═════════════╪══════╪══════════════════╪═════════════╪══════════════╪════════════════╡
│ 0 │ parent_mesh │ 9 │ 3 │ 24576 │ 50688 │ 4913 │
├────┼─────────────┼──────┼──────────────────┼─────────────┼──────────────┼────────────────┤
│ 1 │ ERm │ 24 │ 2 │ 432 │ 648 │ 218 │
├────┼─────────────┼──────┼──────────────────┼─────────────┼──────────────┼────────────────┤
│ 2 │ ER │ 30 │ 3 │ 1296 │ 2808 │ 343 │
├────┼─────────────┼──────┼──────────────────┼─────────────┼──────────────┼────────────────┤
│ 3 │ PM │ 37 │ 2 │ 512 │ 800 │ 289 │
├────┼─────────────┼──────┼──────────────────┼─────────────┼──────────────┼────────────────┤
│ 4 │ Cyto │ 43 │ 3 │ 23280 │ 48312 │ 4788 │
╘════╧═════════════╧══════╧══════════════════╧═════════════╧══════════════╧════════════════╛
If parameter was loaded from xdmf, plot here.
if add_reaction:
visualization.plot(model_cur.pc['j1pulse_fromxdmf'].dolfin_function,
clip_plane=(1, 1, 0), clip_origin=(0.5, 0.5, 0.5))
/usr/lib/python3/dist-packages/smart/visualization.py:167: PyVistaDeprecationWarning: This function is deprecated and will be removed in future version of PyVista. Use vtk with osmesa instead.
pyvista.start_xvfb()
Solving system and storing data#
We create some XDMF files where we will store the output
# Write initial condition(s) to file
results = dict()
result_folder = pathlib.Path("results")
result_folder.mkdir(exist_ok=True)
for species_name, species in model_cur.sc.items:
results[species_name] = d.XDMFFile(
model_cur.mpi_comm_world, str(result_folder / f"{species_name}.xdmf")
)
results[species_name].parameters["flush_output"] = True
results[species_name].write(model_cur.sc[species_name].u["u"], model_cur.t)
We now run the the solver and store the data at each time point to the initialized files. We also integrate A over the cytosolic volume at each time step to monitor the elevation in A over time and we display the concentration of A in the cytosol at t = 0.2 s.
# Set loglevel to warning in order not to pollute notebook output
logger.setLevel(logging.WARNING)
avg_A = [A.initial_condition]
# define integration measure and total volume for computing average A at each time point
dx = d.Measure("dx", domain=model_cur.cc['Cyto'].dolfin_mesh)
volume = d.assemble_mixed(1.0*dx)
# Solve
displayed = False
while model_cur.t < model_cur.final_t:
# Solve the system
model_cur.monolithic_solve()
# Save results for post processing
for species_name, species in model_cur.sc.items:
results[species_name].write(model_cur.sc[species_name].u["u"], model_cur.t)
# compute average A concentration at each time step
int_val = d.assemble_mixed(model_cur.sc['A'].u['u']*dx)
avg_A.append(int_val / volume)
if model_cur.t >= 0.2 and not displayed:
visualization.plot(model_cur.sc['A'].u['u'],
clip_plane=(1, 1, 0), clip_origin=(0.5, 0.5, 0.5))
displayed = True
/usr/lib/python3/dist-packages/smart/visualization.py:167: PyVistaDeprecationWarning: This function is deprecated and will be removed in future version of PyVista. Use vtk with osmesa instead.
pyvista.start_xvfb()
Finally, we plot the average concentration of A over time.
plt.plot(model_cur.tvec, avg_A)
plt.xlabel('Time (s)')
plt.ylabel('Cytosolic concentration of A (μM)')
Text(0, 0.5, 'Cytosolic concentration of A (μM)')
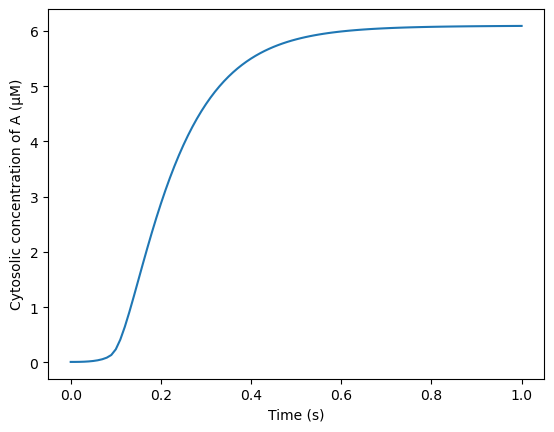
We also compare the AUC for A with previous numerical simulations (regression test).
tvec = np.zeros(len(model_cur.tvec))
for i in range(len(model_cur.tvec)):
tvec[i] = float(model_cur.tvec[i])
auc_cur = np.trapz(np.array(avg_A), tvec)
auc_compare = 4.646230684534995
percent_error = 100*np.abs(auc_cur - auc_compare)/auc_compare
assert percent_error < .01,\
f"Failed regression test: Example 5 results deviate {percent_error:.3f}% from the previous numerical solution"